差分电荷和Bader电荷分布
差分电荷分析(H2O分子为例)
即成键后的电荷密度与对应的成键前的原子电荷密度之差。通过差分电荷密度的计算和分析,可以清楚地得到在成键和成键电子耦合过程中的电荷移动以及成键极化方向等性质。
- 在6x7x8的立方晶格中建立H2O分子结构,并进行结构优化
INCAR参考
1
2
3
4
5
6
7
8
9
10
11
12
13System = relax_H2O
PREC = N
ENCUT = 520
EDIFF = 1e-4
EDIFFG = -0.01
IBRION = 2
ISIF = 2
NSW = 100
NPAR = 4
ISMEAR = 0
SIGMA = 0.05
LCHARG = F
LWAVE = FPOSCAR参考
1
2
3
4
5
6
7
8
9
10
11H2O
1.0
6.0000000000 0.0000000000 0.0000000000
0.0000000000 7.0000000000 0.0000000000
0.0000000000 0.0000000000 8.0000000000
H O
2 1
Direct
0.449999988 0.500000000 0.500000000
0.649999976 0.500000000 0.500000000
0.550000012 0.600000024 0.500000000KPOINTS参考
1
2
3
4
5K-Mesh
0
Monkhorst-Pack
3 3 3
0.0 0.0 0.0
- 提交任务进行计算,完成后得到优化的H2O分子结构(在CONTCAR文件中)
- CONTCAR文件
1
2
3
4
5
6
7
8
9
10
11H2O
1.00000000000000
6.0000000000000000 0.0000000000000000 0.0000000000000000
0.0000000000000000 7.0000000000000000 0.0000000000000000
0.0000000000000000 0.0000000000000000 8.0000000000000000
H O
2 1
Direct
0.4221459669989446 0.5049024206477414 0.5000000000000000
0.6778539970010540 0.5049024206477414 0.5000000000000000
0.5500000119999982 0.5901951827045205 0.5000000000000000
- 建立新文件夹进行差分电荷的计算
首先,新建一个ded的目录并设置相应的INCAR文件,用于计算H2O的电荷密度
1
2
3
4
5
6
7
8
9
10System = relax_H2O
PREC = N
ENCUT = 520
EDIFF = 1e-4
EDIFFG = -0.01
NPAR = 4
ISMEAR = 0
SIGMA = 0.05
LCHARG = T
LWAVE = F即只需要输出CHGCAR文件即可
POSCAR和KPOINTS等与结构优化的一样,其中POSCAR需要使用优化后的CONTCAR
第二部新建立a和b文件夹,分别进行包含1个O原子和2个H原子的电荷密度计算,POSCAR中相应原子位置你不要移动并保持INCAR与计算H2O分子时候的一致
3个独立的计算完成后,会分别在ded文件夹、a文件夹和b文件夹下产生CHGCAR文件夹,此时需要将a和b文件夹下的CHGCAR文件重命名为.vasp格式,可以分别命名为a.vasp和b.vasp
将H2O的电荷密度CHGCAR拖到VESTA软件中,显示

- 依次点击Edit-> Edit Data-> Volumetric data -> Import,分别导入a.vasp和b.vasp,即相应原子的电荷密度
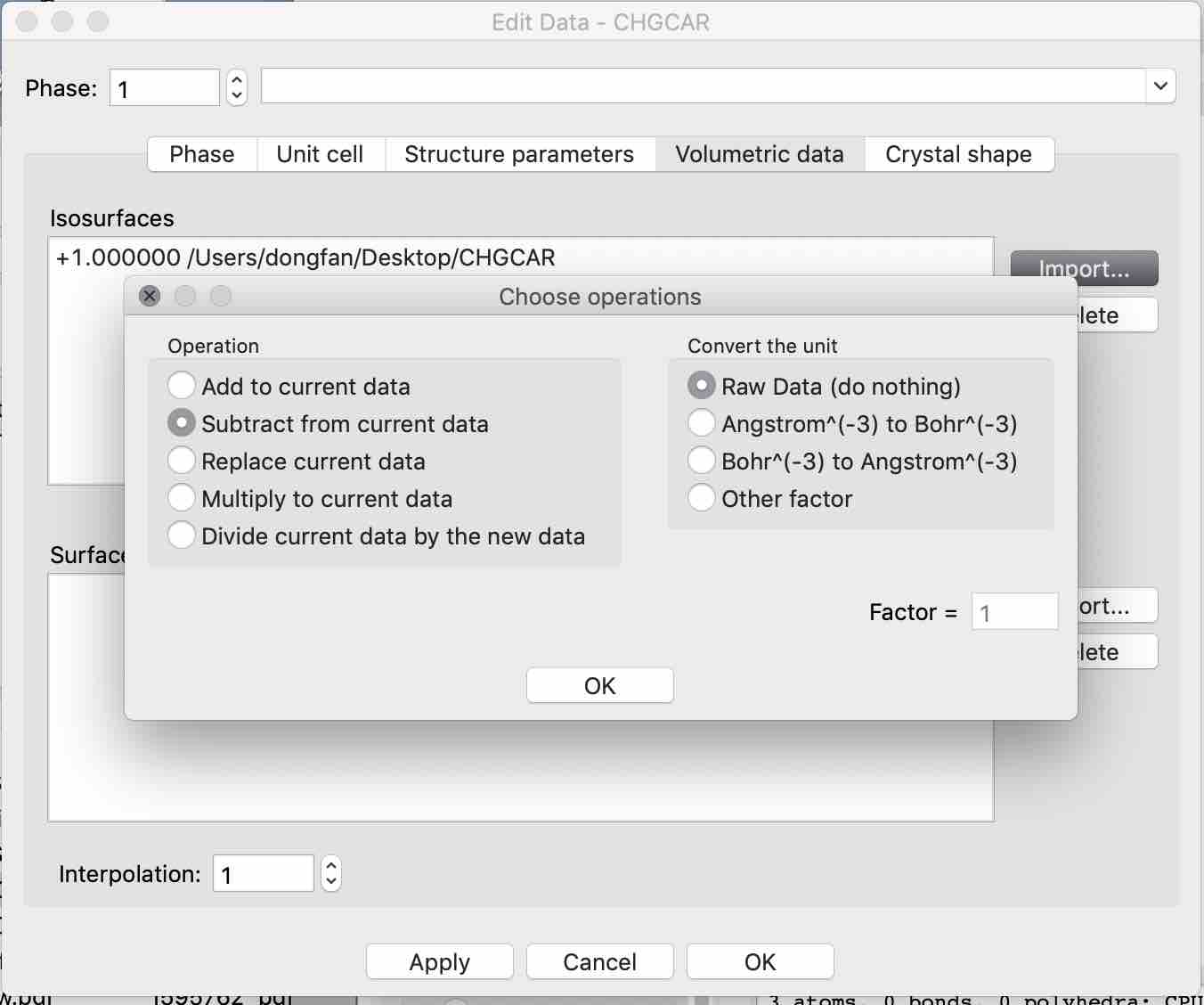
- 注意这里需要选择Subtract from current data
- 点击ok,显示

Bader电荷分析(H2O分子为例)
- 在6x7x8的立方晶格中建立H2O分子结构,并进行结构优化
INCAR参考
1
2
3
4
5
6
7
8
9
10
11
12
13System = relax_H2O
PREC = N
ENCUT = 520
EDIFF = 1e-4
EDIFFG = -0.01
IBRION = 2
ISIF = 2
NSW = 100
NPAR = 4
ISMEAR = 0
SIGMA = 0.05
LCHARG = F
LWAVE = FPOSCAR参考
1
2
3
4
5
6
7
8
9
10
11H2O
1.0
6.0000000000 0.0000000000 0.0000000000
0.0000000000 7.0000000000 0.0000000000
0.0000000000 0.0000000000 8.0000000000
H O
2 1
Direct
0.449999988 0.500000000 0.500000000
0.649999976 0.500000000 0.500000000
0.550000012 0.600000024 0.500000000KPOINTS参考
1
2
3
4
5K-Mesh
0
Monkhorst-Pack
3 3 3
0.0 0.0 0.0
- 提交任务进行计算,完成后得到优化的H2O分子结构(在CONTCAR文件中)
- CONTCAR文件
1
2
3
4
5
6
7
8
9
10
11H2O
1.00000000000000
6.0000000000000000 0.0000000000000000 0.0000000000000000
0.0000000000000000 7.0000000000000000 0.0000000000000000
0.0000000000000000 0.0000000000000000 8.0000000000000000
H O
2 1
Direct
0.4221459669989446 0.5049024206477414 0.5000000000000000
0.6778539970010540 0.5049024206477414 0.5000000000000000
0.5500000119999982 0.5901951827045205 0.5000000000000000
- 建立新文件夹进行Bader电荷的计算,这里新建一个Bader的目录并设置相应的INCAR文件
INCAR参考
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15System = bader
PREC = N
ENCUT = 520
EDIFF = 1e-4
EDIFFG = -0.02
ISMEAR = 0;
SIGMA = 0.05
LCHARG = T
LWAVE = F
NPAR = 4
LAECHG =.TRUE.
LCHARG =.TRUE.
NGX= 99 ###取值太大,可以只加倍
NGY= 117 ###取值太大,可以只加倍
NGZ= 135 ###取值太大,可以只加倍注:INCAR中的参数需要保证足够密,可以在结构优化后,使用
grep -A3 'NGX' OUTCAR命令获得结构优化中所使用的参数,随后在Bader计算中进行加密即可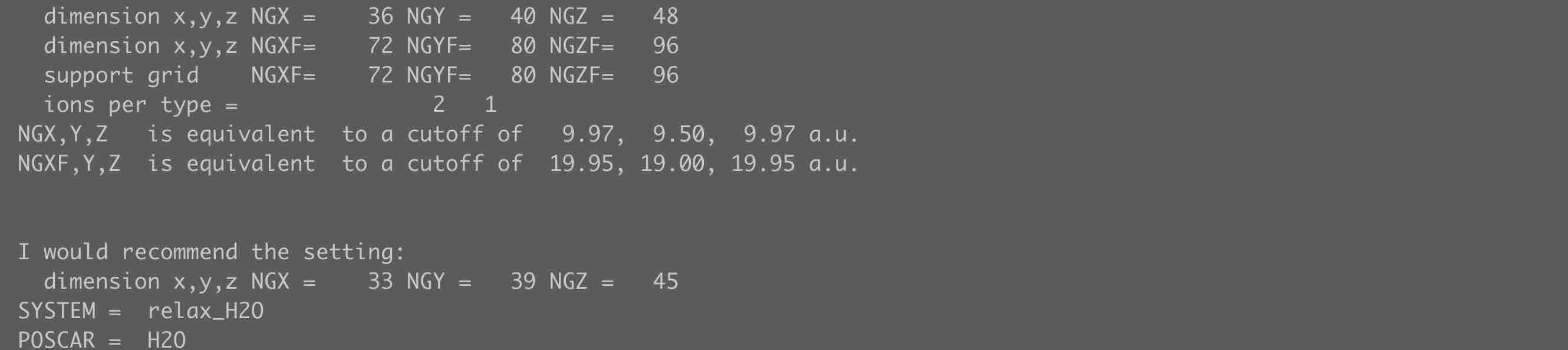
- 计算完成后,成功写入了AECCAR0和AECCAR0文件,接下来在终端运行以下2行命令
1
2
3
4chgsum.pl AECCAR0 AECCAR2
bader CHGCAR -ref CHGCAR_sum
#在第二个命令报错的时候运行以下备用命令
bader -b weight CHGCAR -ref CHGCAR_sum
- 计算得到的电荷转移数据储存于ACD.dat文件中
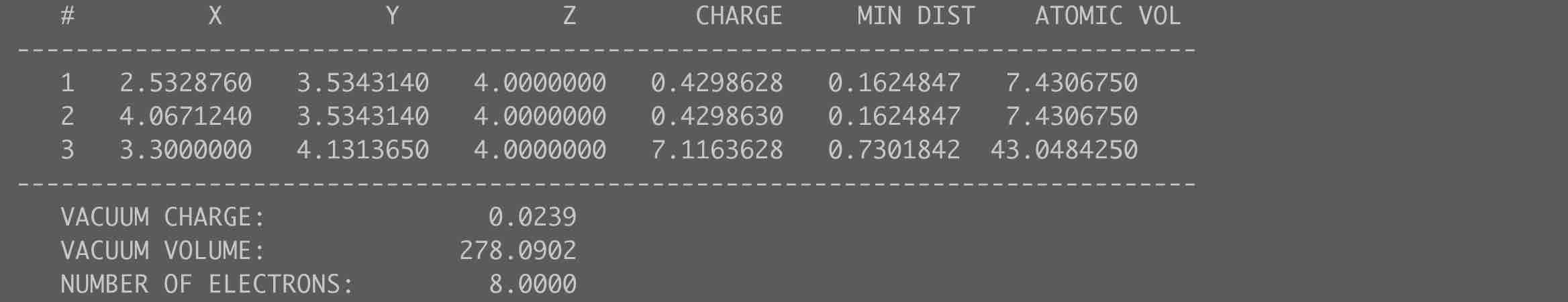
- 其中原子顺序于POSCAR中的一致,每个相应的原子的价电子减去CHARGE这一列的数值代表电荷转移的数值
- 减掉以后为负数代表电子向该原子转移,正数代表该原子失去电子
- 因此,水分子中的电荷分别为H:0.57,O:-1.12
- 与文献中采用Gaussian计算的结果进行比较
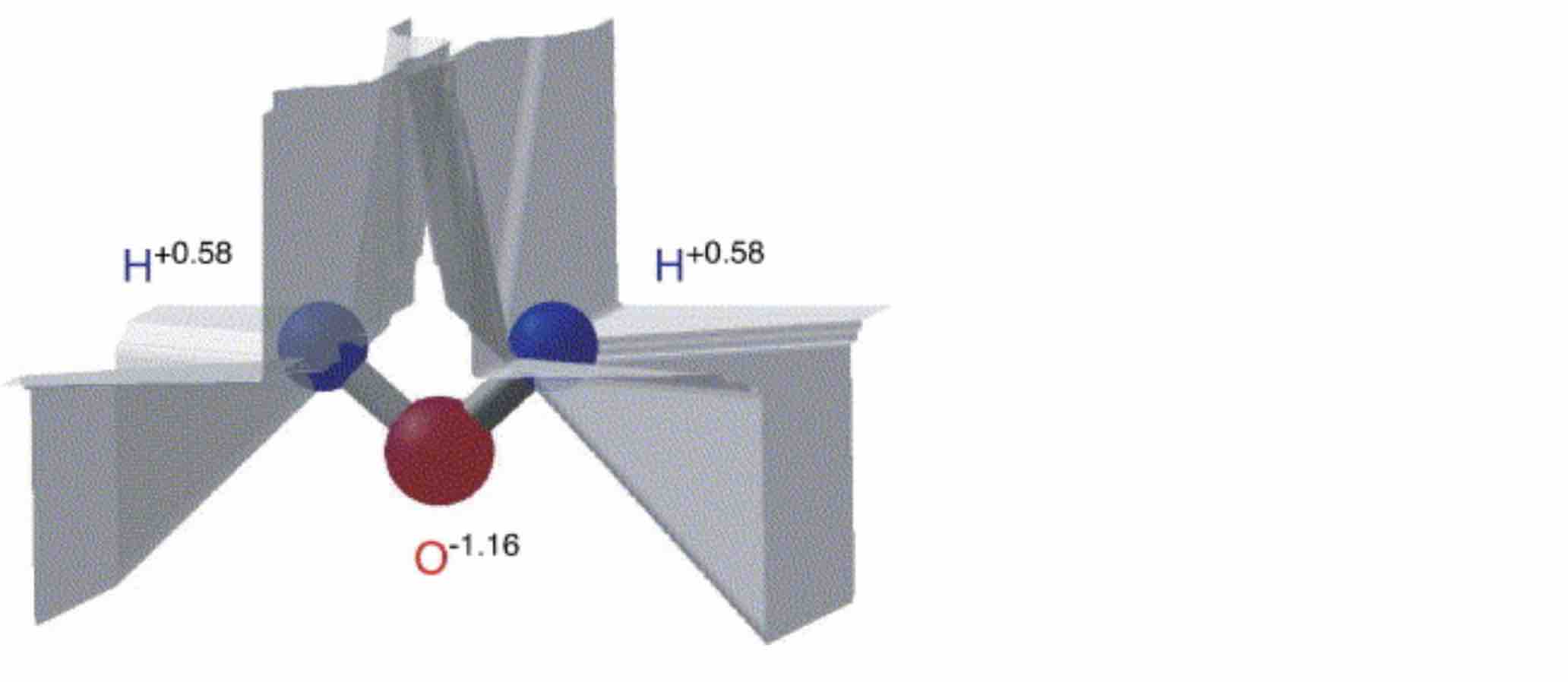
- 与文献符合的很好
References
- Blog Link: http://agrh.github.io/2019/08/06/ded/
- Copyright Declaration: The author owns the copyright, please indicate the source reproduced.