过渡态理论以及Ammonia flipping计算实例
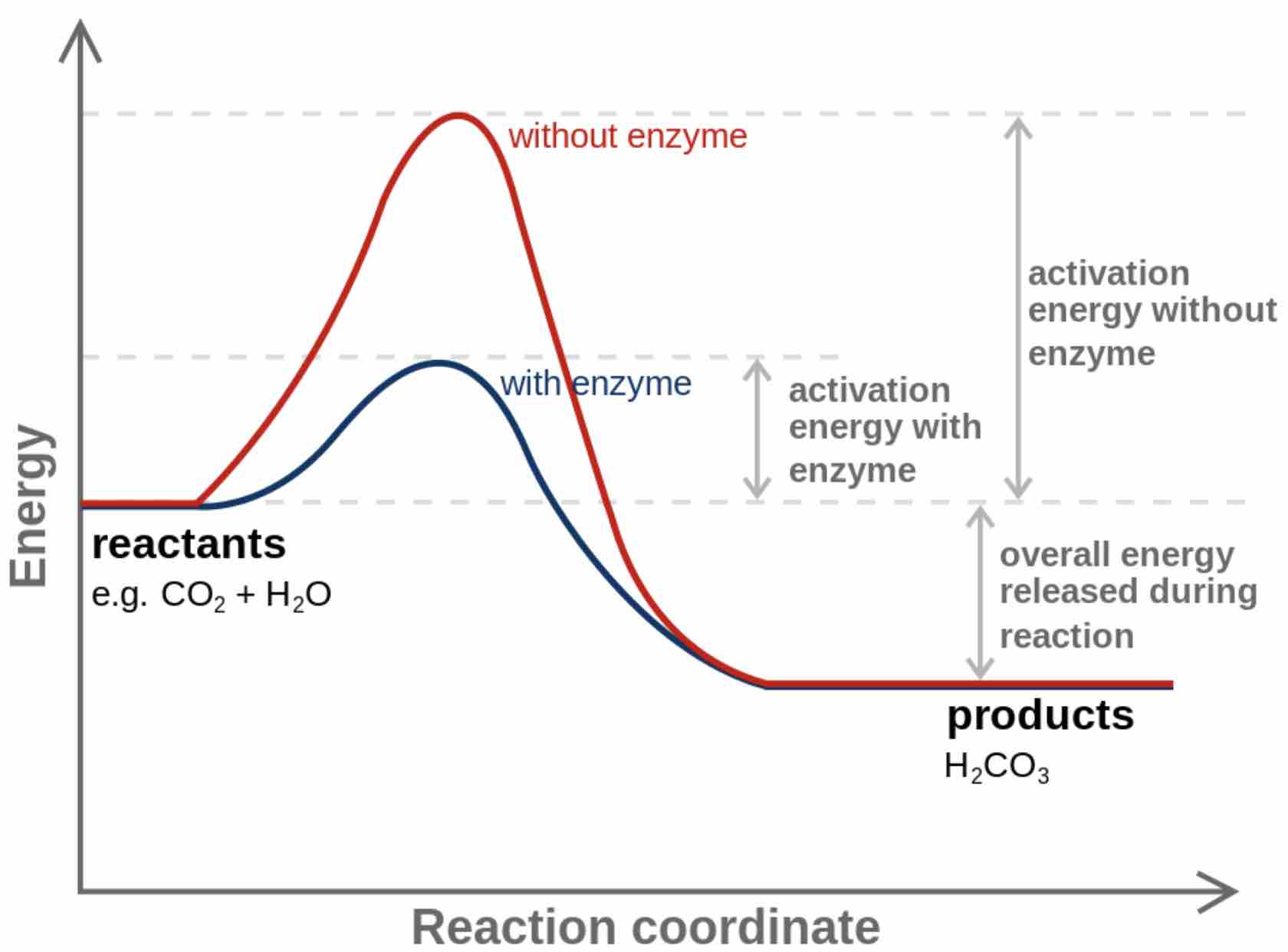
- 当体系处于反应物或者产物的时候,整个体系为平衡态
- 从反应物到产物的过程中至少有一个能量最高点,该点能量与反应物的差值即为反应所需要跨越的势垒,其活化能
- 反应路径至少一条
Ammonia flipping
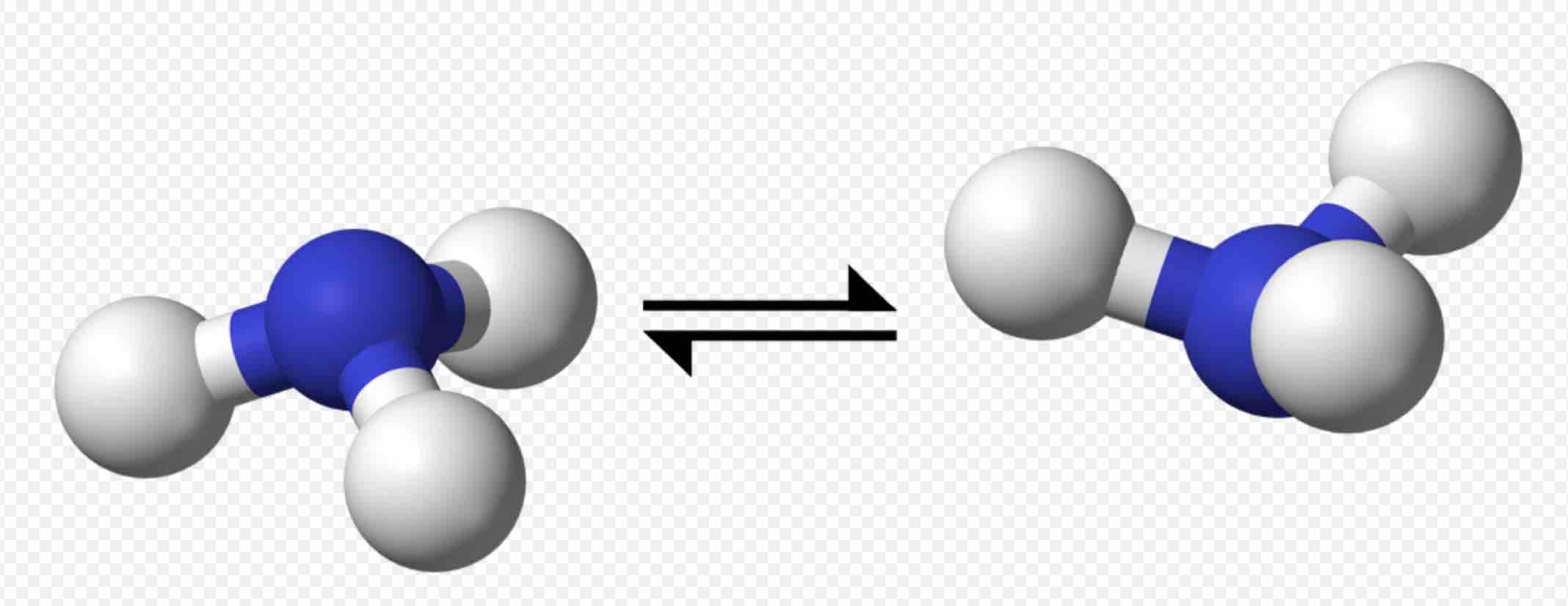
官方例子可以参考这里.
这里我们采用与VASP官方不一样的climbing image nudged elastic band (CI-NEB)的方法计算Ammonia flipping反应的过渡态。
1. 首先进行初态结构和末态结构的优化,采用PBE泛函,在6x7x8的晶格中放入一个NH3分子,采用3x3x3的k-points。具体如下:
分别建立ini和fin的文件夹,进行初态和末态的结构优化工作,其中均INCAR为:
1
2
3
4
5
6
7
8
9
10
11
12
13System = relax_diamond
PREC = N
ENCUT = 500
EDIFF = 1e-4
EDIFFG = -0.01
IBRION = 2
ISIF = 2
NSW = 100
NPAR = 4
ISMEAR = 0
SIGMA = 0.05
LCHARG = F
LWAVE = FKPOINTS 为:
1
2
3
4
5K-Mesh
0
Monkhorst-Pack
3 3 3
0.0 0.0 0.0ini文件夹中POSCAR为:
1
2
3
4
5
6
7
8
9
10
11
12
13ammonia flipping-ini
1.00000000000000
6.000000 0.000000 0.000000
0.000000 7.000000 0.000000
0.000000 0.000000 8.000000
H N
3 1
Selective
Direct
0.636428 0.567457 0.5491645 T T T
0.500000 0.364985 0.5491330 T T T
0.363572 0.567457 0.5491645 T T T
0.500000 0.500000 0.5000000 F F Ffin文件夹中POSCAR为:
1
2
3
4
5
6
7
8
9
10
11
12
13ammonia flipping-fin
1.00000000000000
6.000000 0.000000 0.000000
0.000000 7.000000 0.000000
0.000000 0.000000 8.000000
H N
3 1
Selective
Direct
0.636428 0.567457 0.4508355 T T T
0.500000 0.364985 0.4508670 T T T
0.363572 0.567457 0.4508355 T T T
0.500000 0.500000 0.5000000 F F FPOTCAR采用cat命令或者vpot.py自行添加。提交任务的pbs脚本现在就开始统一为重新编译过的neb脚本,路径
~/fd/script/fd_neb.sh重新编译过得VASP版本包括了VTST脚本,同时后续所有neb相关计算均依赖与该脚本
最后分别提交任务,结构优化完成后
cp CONTCAR POSCAR优完成后的初态:
1
2
3
4
5
6
7
8
9
10
11
12
13ammonia flipping-ini
1.00000000000000
6.0000000000000000 0.0000000000000000 0.0000000000000000
0.0000000000000000 7.0000000000000000 0.0000000000000000
0.0000000000000000 0.0000000000000000 8.0000000000000000
H N
3 1
Selective dynamics
Direct
0.6362235817250829 0.5674490901961542 0.5488487003974116 T T T
0.5000000000000000 0.3651050627569393 0.5487383170743516 T T T
0.3637764182749170 0.5674490901961542 0.5488487003974116 T T T
0.5000000000000000 0.5000000000000000 0.5000000000000000 F F F优化完成后的末态:
1
2
3
4
5
6
7
8
9
10
11
12
13ammonia flipping-fin
1.00000000000000
6.0000000000000000 0.0000000000000000 0.0000000000000000
0.0000000000000000 7.0000000000000000 0.0000000000000000
0.0000000000000000 0.0000000000000000 8.0000000000000000
H N
3 1
Selective dynamics
Direct
0.6362237799950755 0.5674491584408682 0.4511512564865826 T T T
0.5000000000000000 0.3651049239616792 0.4512616320948187 T T T
0.3637762200049245 0.5674491584408682 0.4511512564865826 T T T
0.5000000000000000 0.5000000000000000 0.5000000000000000 F F Fk可以看到未固定原子位置的3个氢原子均进行了不同程度的优化
2. 使用nebmake.pl命令生成中间态结构
在工作目录下(该目录下包含ini和fin的文件夹)执行以下命令:nebmake.pl ini/CONTCAR fin/CONTCAR 6,即表示会在初态和末态之间插入6个中间态结构来进行过渡态的计算,同时会在工作目录产生00-07的文件夹。
在工作目录添加用于计算过渡态的INCAR文件,参考:
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21System = Ammonia flipping
PREC = N
ENCUT = 500
EDIFF = 1e-4
EDIFFG =-0.02
KSPACING= 0.5
ISIF = 2
NSW = 500
ISMEAR = 0;
SIGMA = 0.05
LCHARG = FALSE
LWAVE = FALSE
ISTART = 0
#NEB 以下为计算NEB参数
IBRION = 3
POTIM = 0
IOPT = 3
ICHAIN = 0
LCLIMB = .TRUE.
SPRING =-5
IMAGES =6 #所插点数目必须能够被CPU整除最后在工作目录添加与结构优化中相同的KPOINTS、POTCAR以及neb脚本文件,最后提交计算任务。
计算过程中可以使用nebefs.pl检查力的收敛情况。
计算收敛后,vasp.log文件尾部会出现
reached required accuracy - stopping structural energy minimisation字样,证明neb计算完成。如在设定的迭代步数内仍未收敛,则需要分别将01-06文件夹里面的CONTCAR文件拷贝为POSCAR文件,继续优化,直至收敛。
3. 计算完成
计算完成后,直接使用nebresult.pl进行结构后处理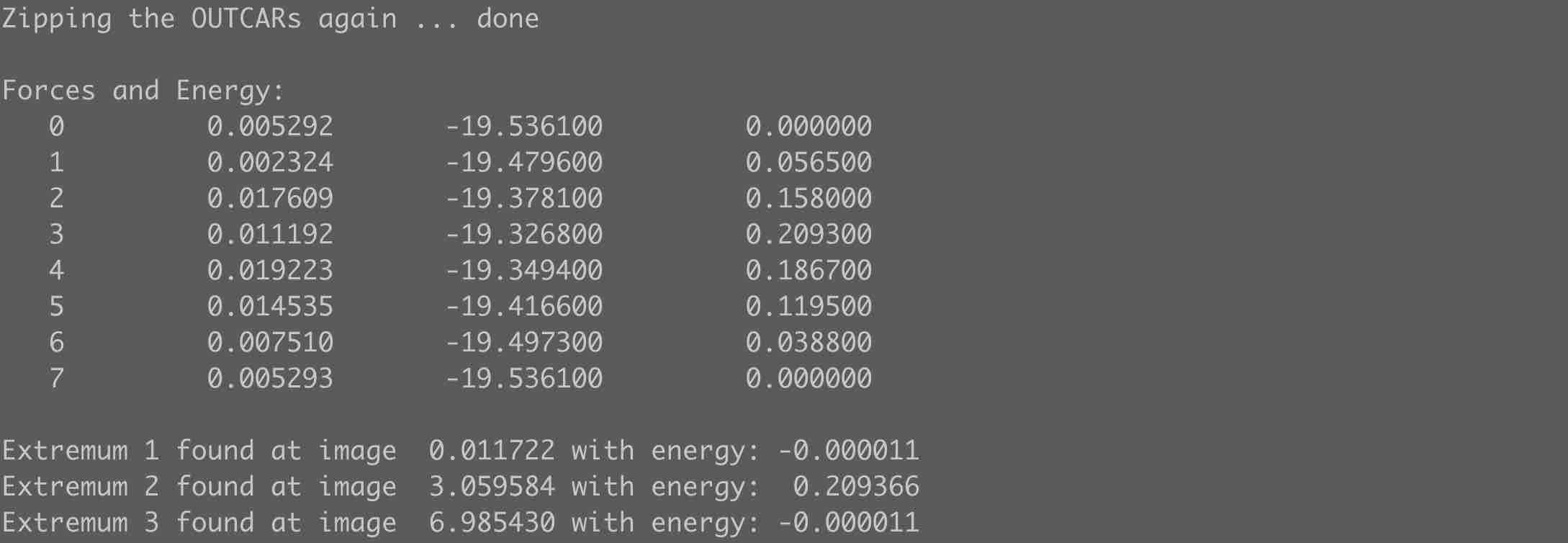
最终,我们会得到mep.ps的文件可以直接使用ps或者其他绘图软件打开。原始的数据文件储存于neb.dat文件,也可以使用origin等绘图软件绘制。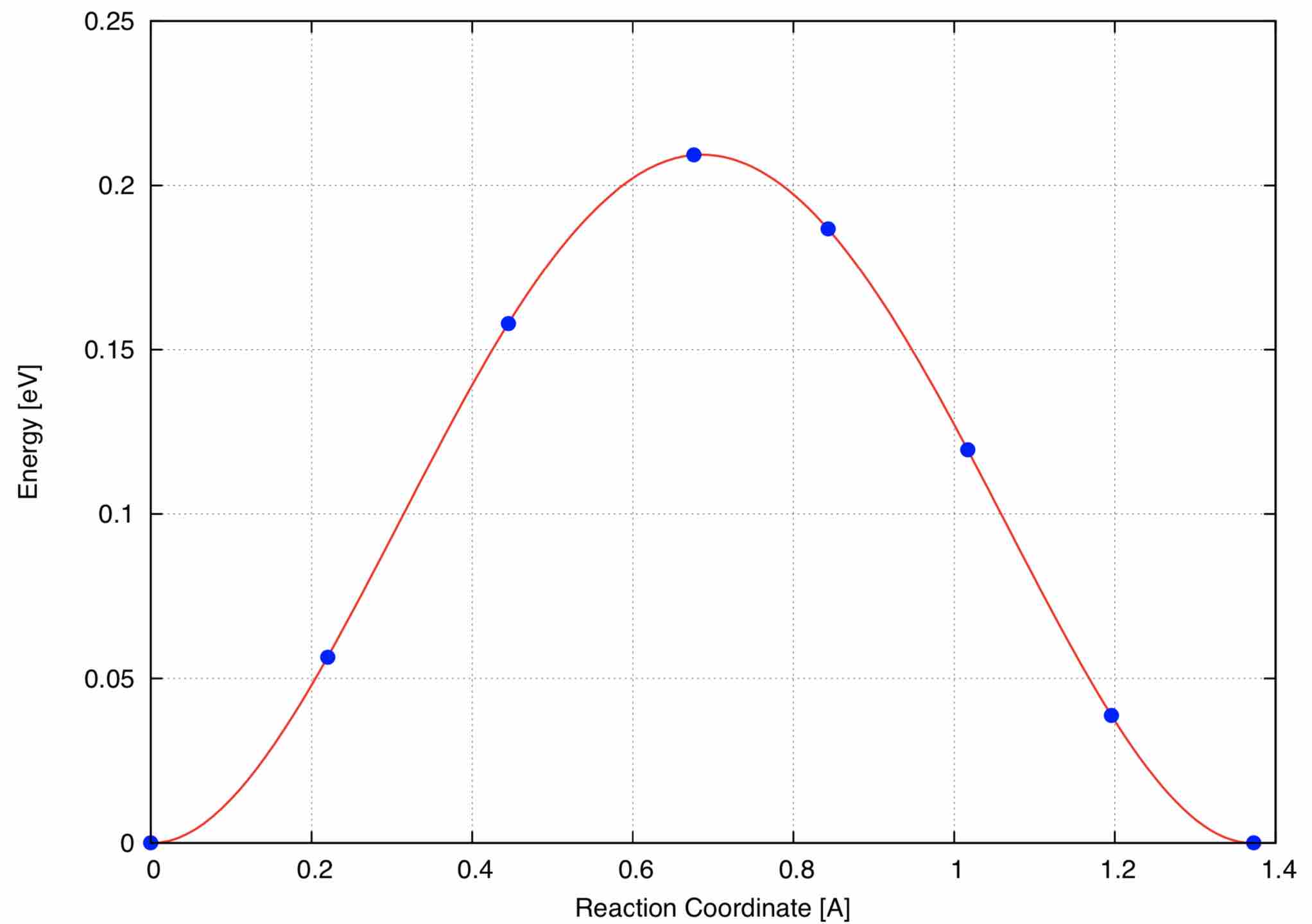
可以看到采用该方法计算得到的势垒为0.2094 eV,与官方实例得到的0.2073 eV仅仅相差0.002,同时我们采用的计算精度也比VASP官方更加精确。
<完>
References
[1] https://en.wikibooks.org/wiki/Structural_Biochemistry/Enzyme/Transition_state
[2] https://en.wikibooks.org/wiki/Structural_Biochemistry/Enzyme/Activation_energy
[3] https://www.vasp.at/vasp-workshop/tutorials/tutorial_ammonia_flipping.pdf
[4] https://www.youtube.com/watch?v=hHv4M-Lm_fw
- Blog Link: http://agrh.github.io/2019/08/01/neb/
- Copyright Declaration: The author owns the copyright, please indicate the source reproduced.