金刚石(100)表面电子结构计算
关于半导体表面重构的原理可以参考这一篇文献,讲的非常详细和基础。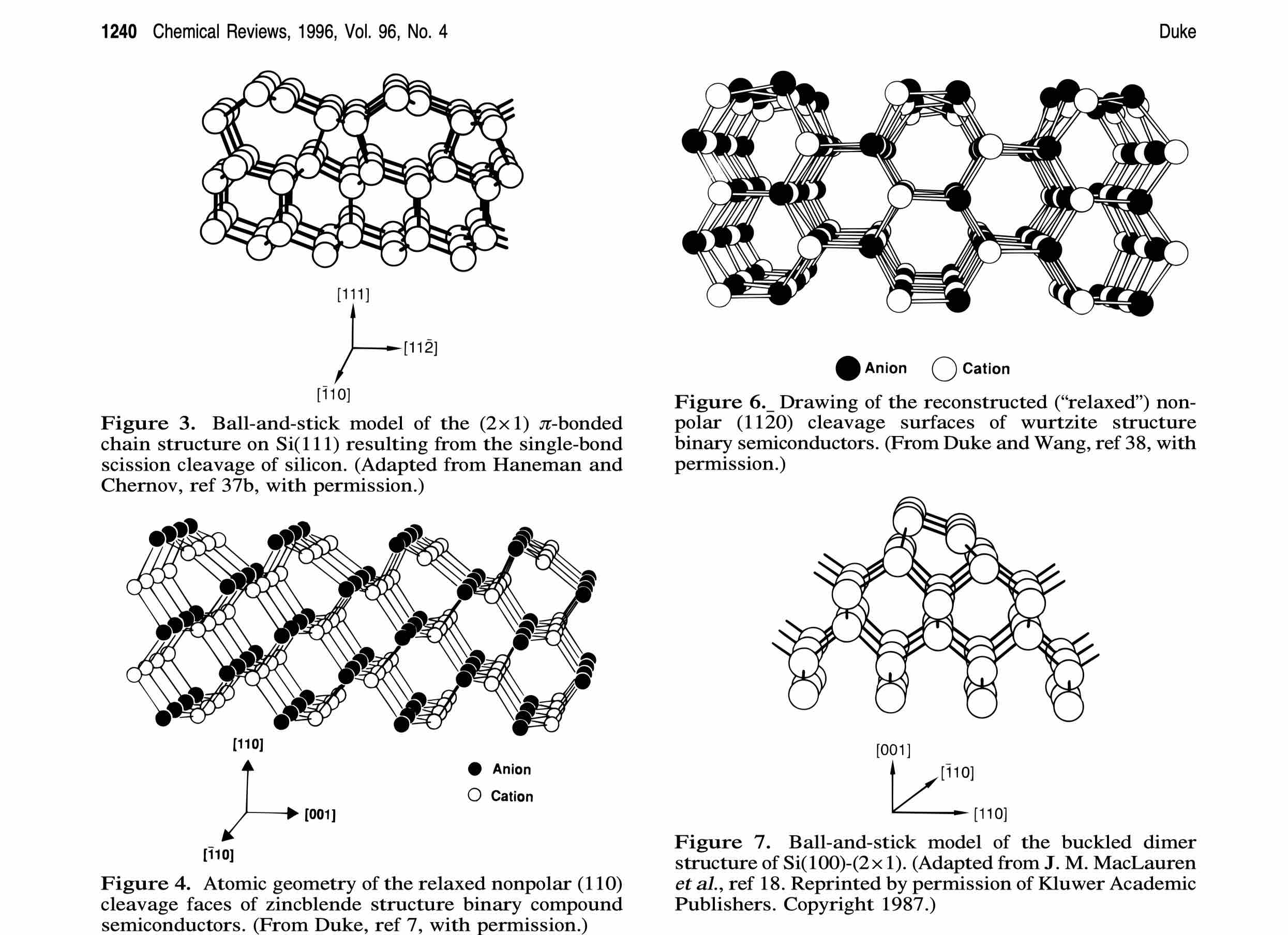
1. 获得体相金刚石结构并切割金刚石2x1-(100)表面,参考上一节内容
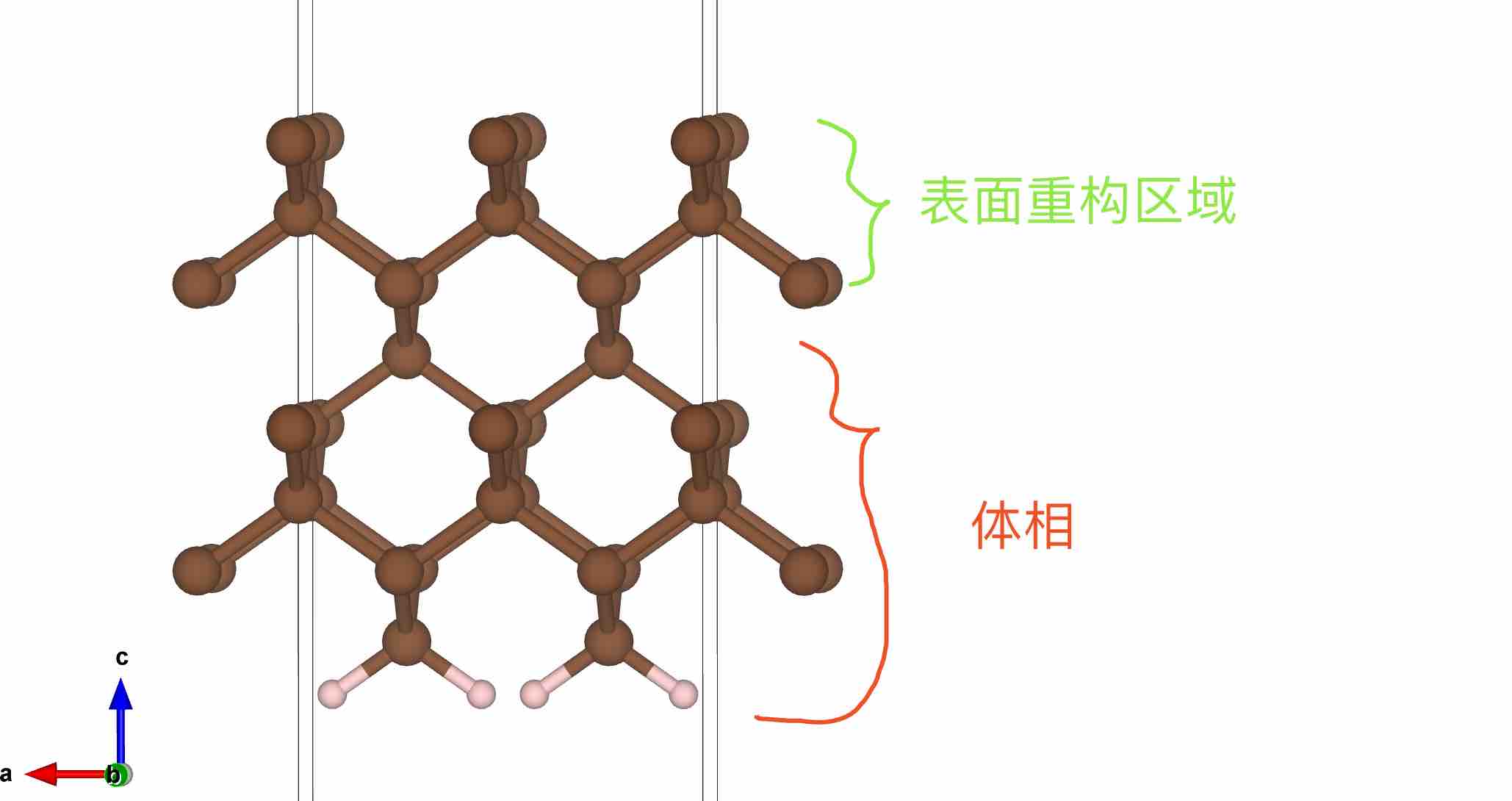
2. 此时,MS自动氢饱和后的C-H键长和键角并不是最合理的,因此需要首先进行H原子的结构弛豫
此时POSCAR文件为:
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29C_mp-66_(diamond_100)
1.0
5.0539999008 0.0000000000 0.0000000000
0.0000000000 2.5269999504 0.0000000000
0.0000000000 0.0000000000 20.7210998535
C H
16 4
Selective Dynamics
Direct
0.250000000 0.500000000 0.344929993 F F F
0.250000000 0.500000000 0.172470003 F F F
0.000000000 0.000000000 0.431169987 F F F
0.000000000 0.000000000 0.258700013 F F F
0.250000000 0.000000000 0.388049990 F F F
0.250000000 0.000000000 0.215580001 F F F
0.000000000 0.500000000 0.474280000 F F F
0.000000000 0.500000000 0.301820010 F F F
0.750000000 0.500000000 0.344929993 F F F
0.750000000 0.500000000 0.172470003 F F F
0.500000000 0.000000000 0.431169987 F F F
0.500000000 0.000000000 0.258700013 F F F
0.750000000 0.000000000 0.388049990 F F F
0.750000000 0.000000000 0.215580001 F F F
0.500000000 0.500000000 0.474280000 F F F
0.500000000 0.500000000 0.301820010 F F F
0.065830000 0.500000000 0.140699998 T T T
0.434170008 0.500000000 0.140699998 T T T
0.565829992 0.500000000 0.140699998 T T T
0.934170008 0.500000000 0.140699998 T T TINCAR设置与之前结构优化一致,KPOINTS设置为
4 8 1,POTCAR使用PBE类型的C和H:
grep TI POTCAR
3. 计算完成后将CONTCAR文件拷贝为POSCAR文件,并修改为
1 | C_mp-66_(diamond_100) |
- 真空层厚度大约为13.7 Å。这一步主要目的是将体相部分原子固定,随后优化表面态,其中最表面处的原子需要手动调节
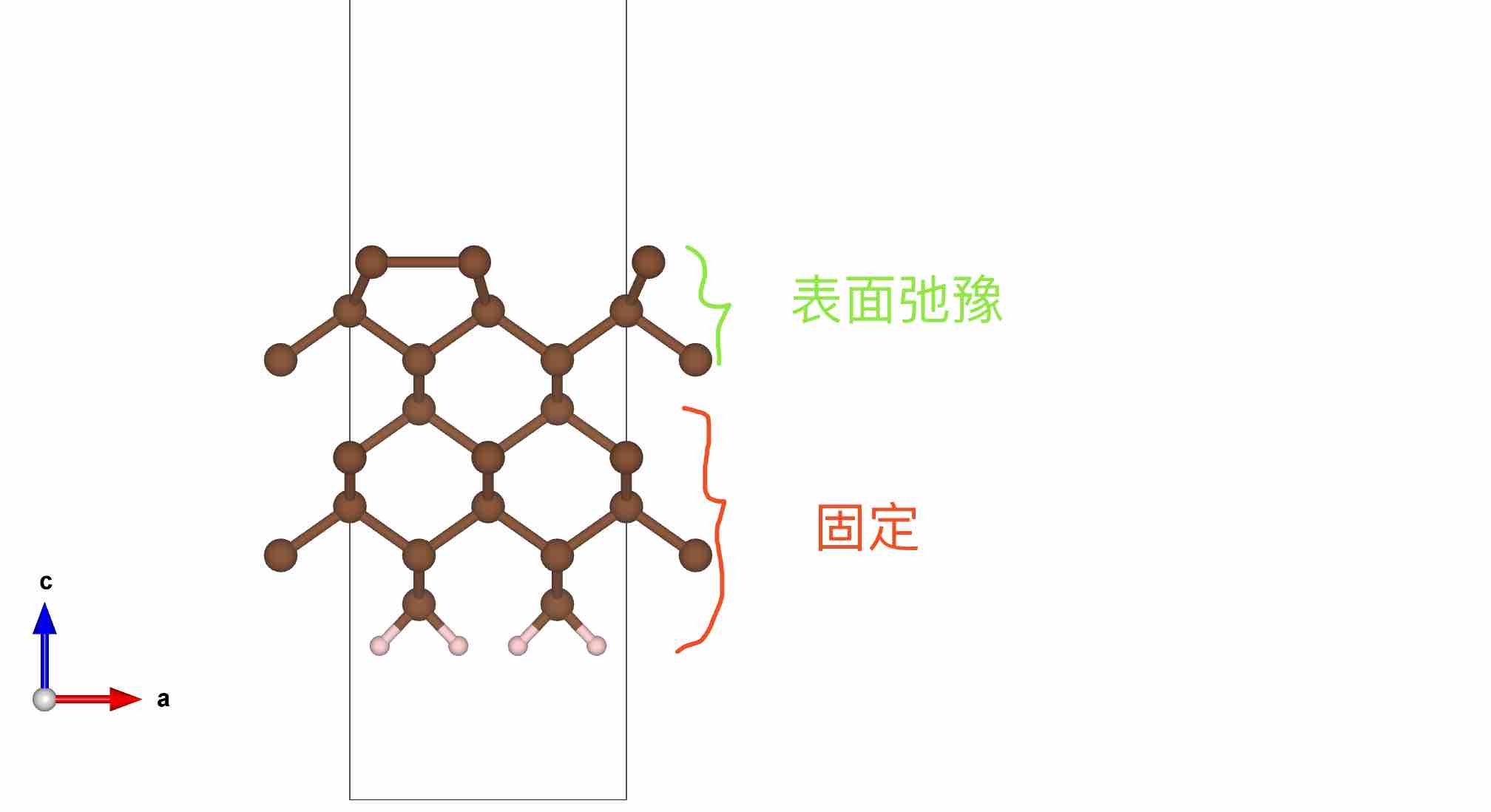
4. 计算完成后将CONTCAR文件拷贝为POSCAR文件。此后,所有物理性质计算和分析均以此POSCAR为准,不再变动
- 优化完成后结构
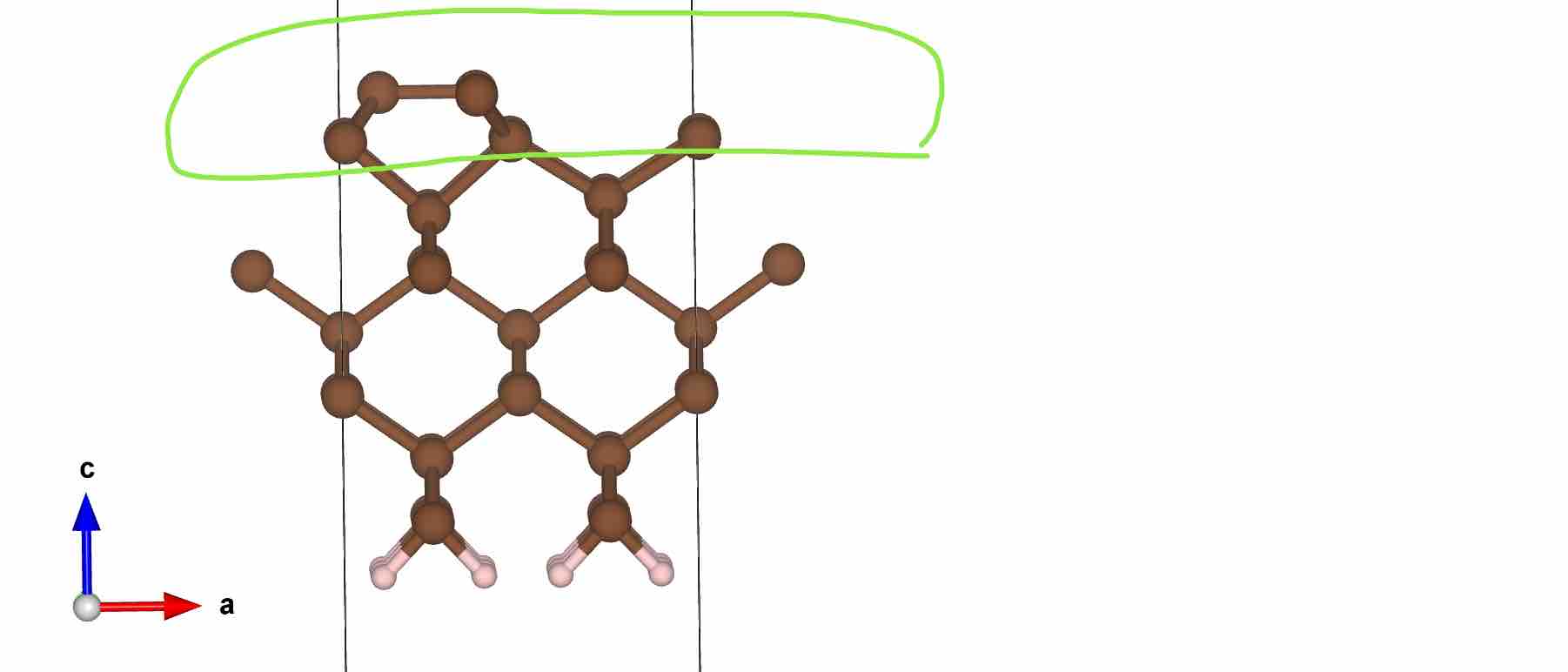
- 检查
OUTCAR中原子受力情况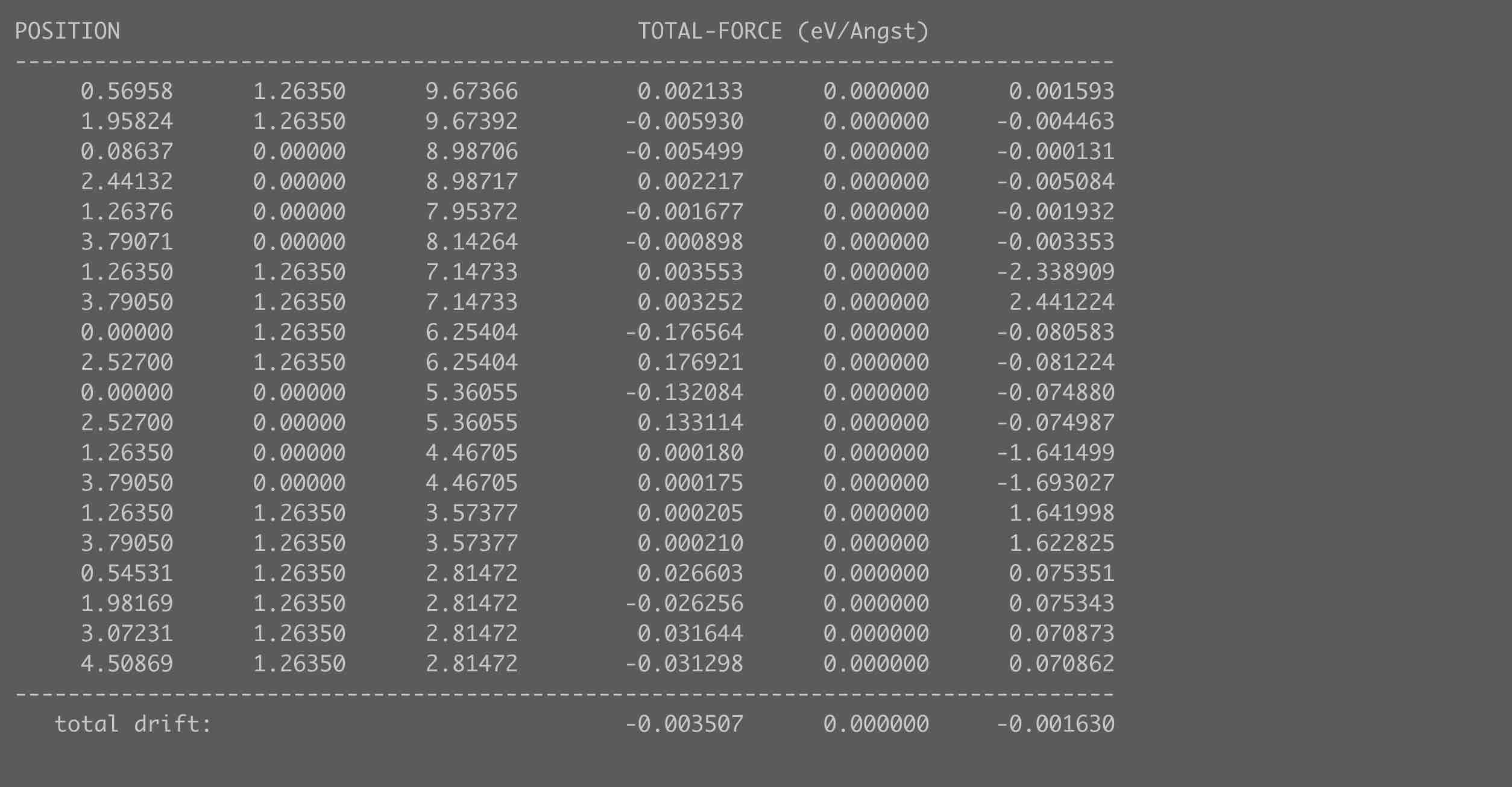
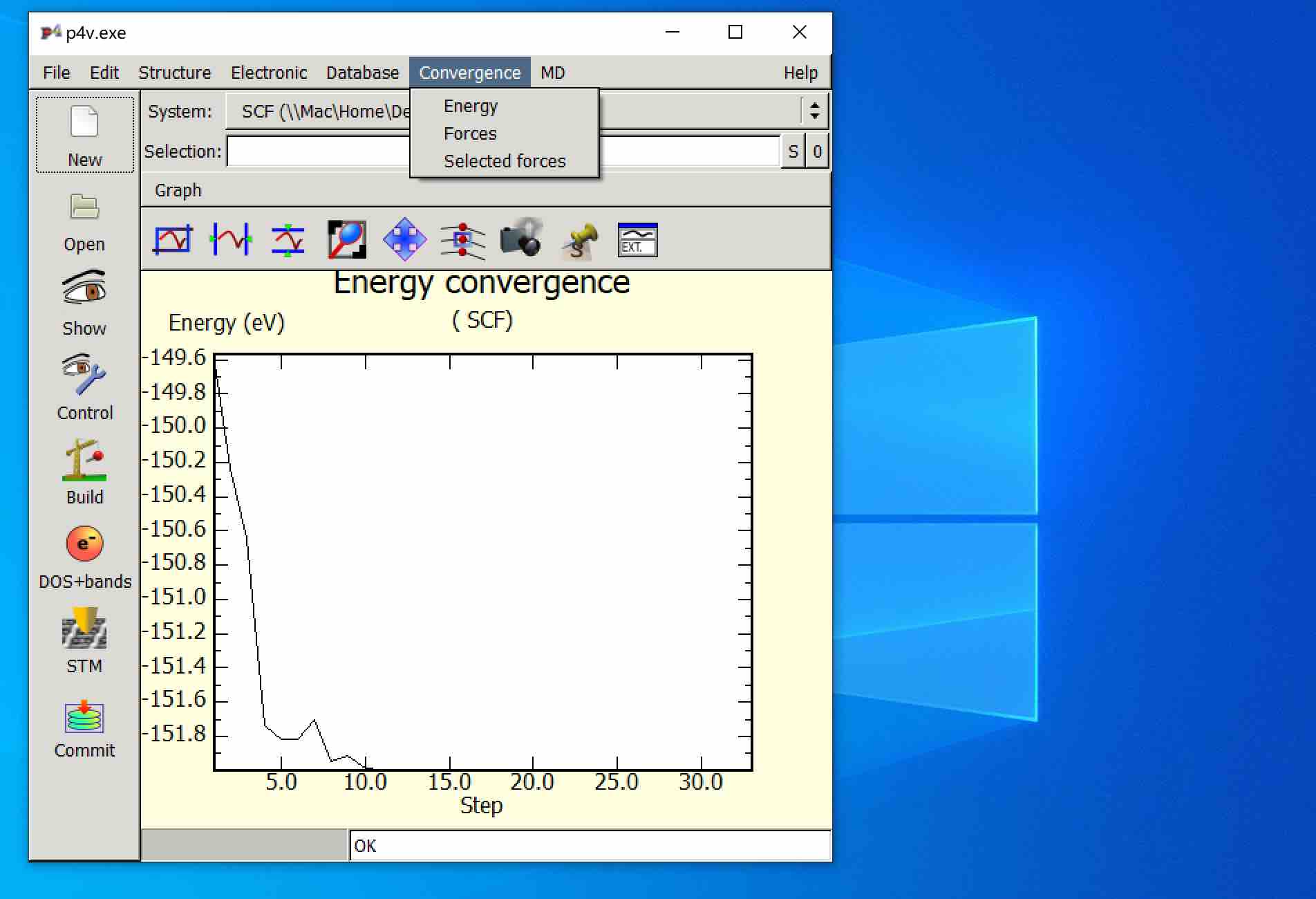
- 也可以在P4VASP中检查可视化原子收敛情况
4. 新建文件夹进行表面能带和态密度的计算,参考上一节内容
- 需要注意表面结构的能带计算中
KPOINTS路径的选择,比体相材料的路径选择简单,因为在相当于只需要在二维布里渊区路径范围内采样。可以参考二维材料的路径选择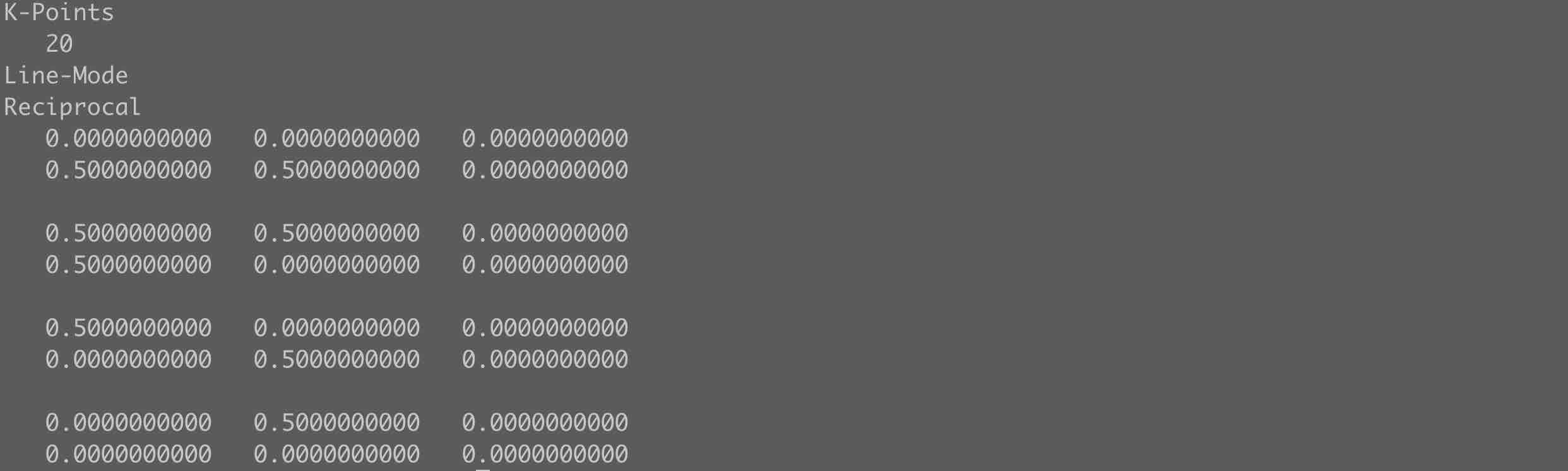
- 计算得到的diamond-2x1(100)表面能带
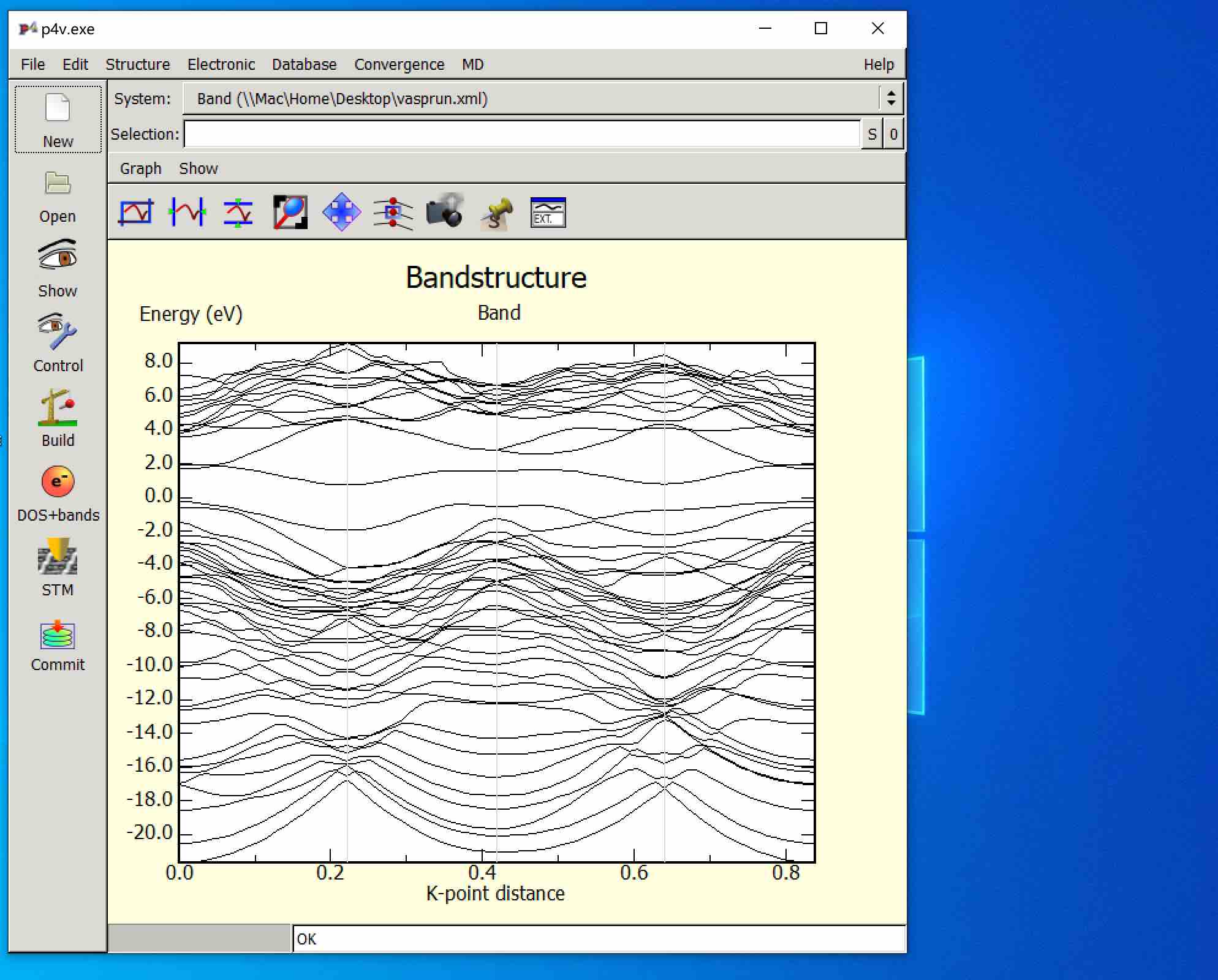
5. Homework
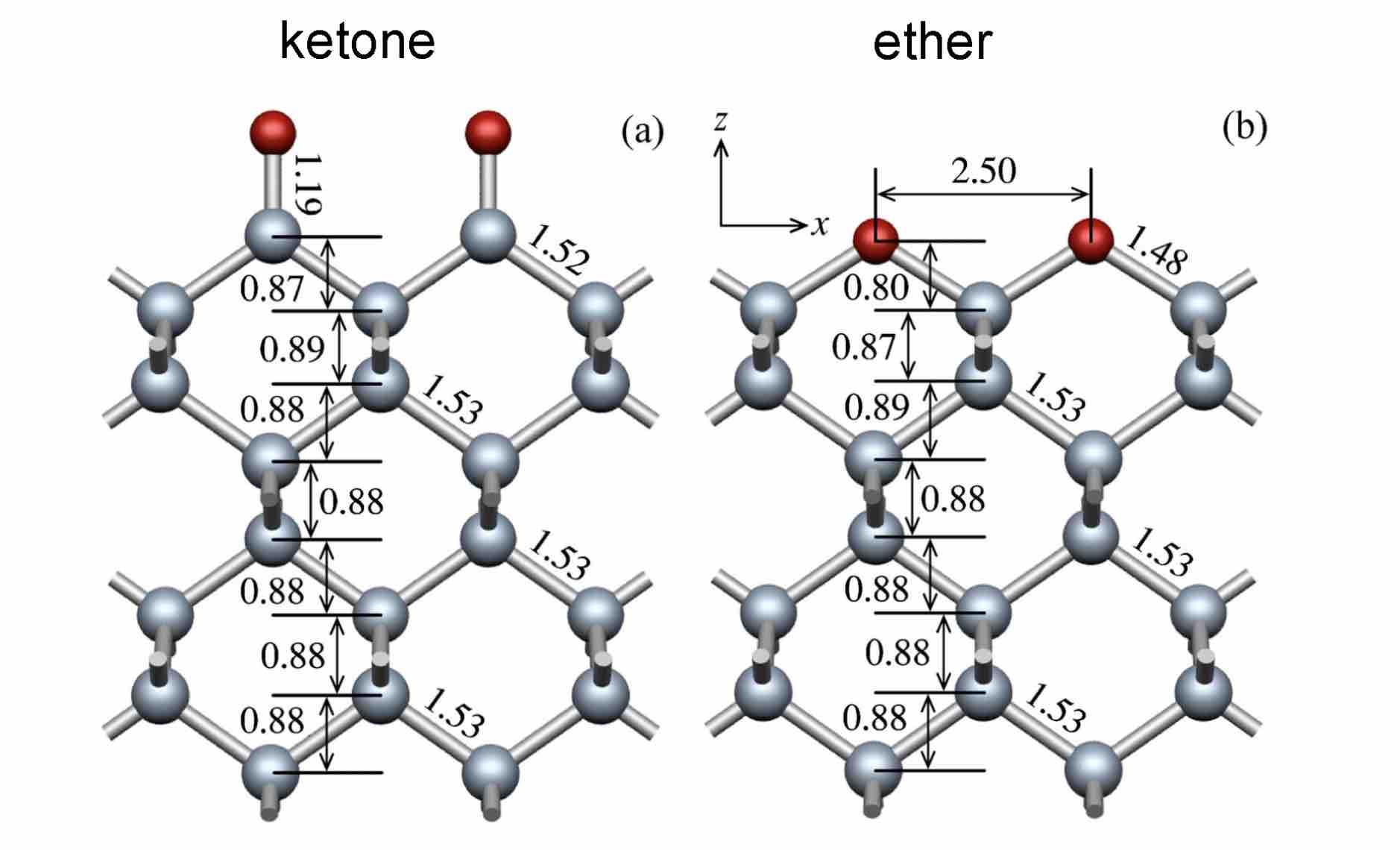
- 计算氧化金刚石(100)表面的能带和态密度曲线,并做初步理论分析。氧化模型分为Ether和Ketone两种。参考以下文献
References
[1] VASP documentation
[2] Chem. Rev. 1996, 96, 1237−1259
[3] Phys. Rev. B 73, 2006, 085313
- Blog Link: http://agrh.github.io/2019/07/30/surface-dia/
- Copyright Declaration: The author owns the copyright, please indicate the source reproduced.