Calculate the band structure and density of states of diamond with the VASP code
*Note: This tutorial is for beginners only.
In the previous chapter, we have completed the structure optimization of diamond and obtained the optimized diamond crystal structure using the PBE functional. In this section, we will complete the calculation of diamond band and electron state density.
>> Create a new folder (name: scf) and perpar four input files and script for submitting task under it
INCAR:
1
2
3
4
5
6
7
8
9
10
11
12
13System = SCF
PREC = N
ISTART = 0
ICHARG =2
ENCUT = 520
EDIFF = 1e-5
EDIFFG = -0.001
IBRION = -1
ISMEAR = 0
SIGMA = 0.05
LCHARG = T
LWAVE = T
NPAR = 4KPOINTS (Please make sure that k points are dense enough!):
1
2
3
4
5K-Mesh
0
Monkhorst-Pack
11 11 11
0.0 0.0 0.0POTCAR:
Copy it directly from the folder. The head of POTCAR of C:
- POSCAR:
1
2
3
4
5
6
7
8
9
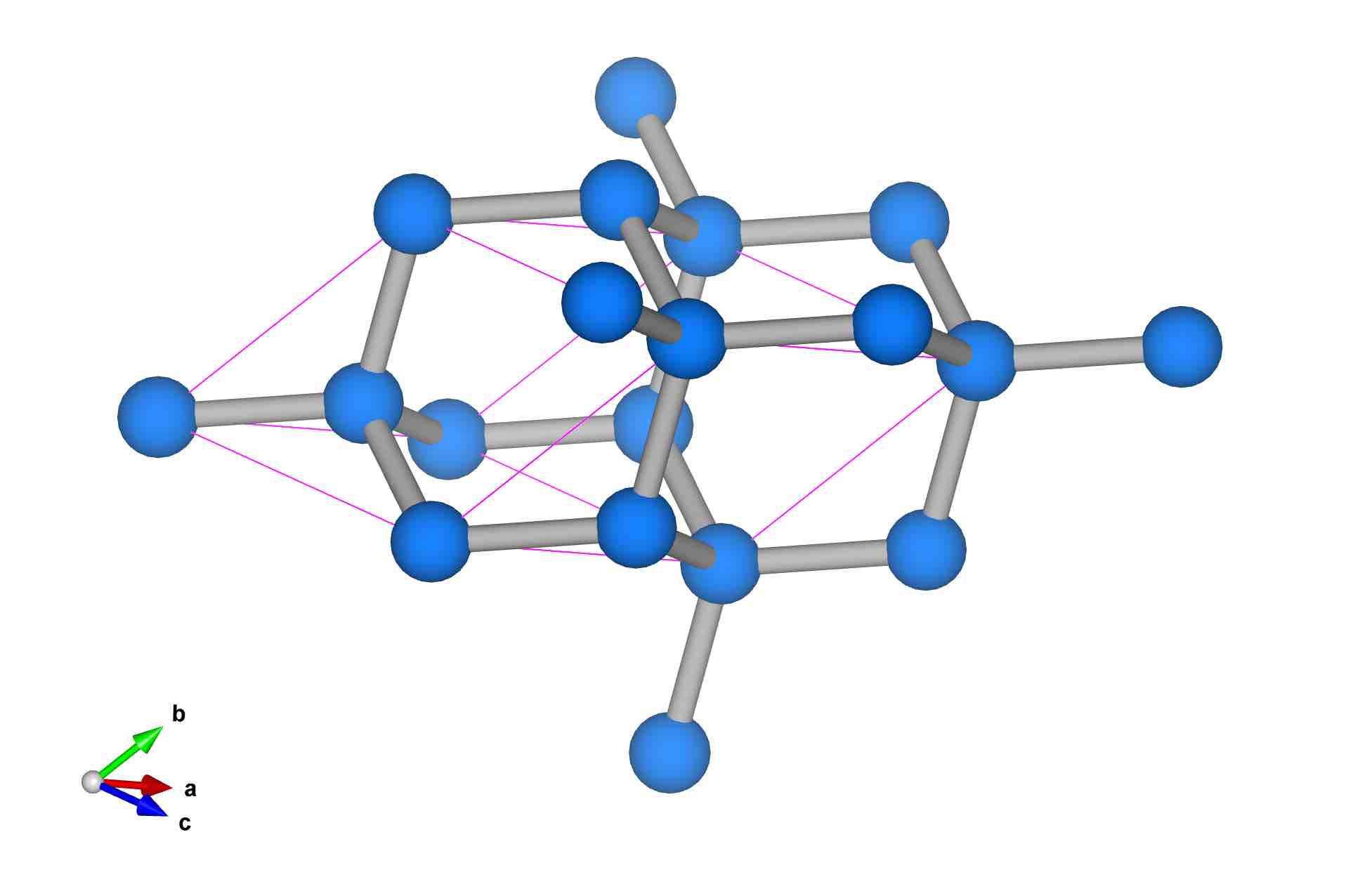
10Diamond_Primitive Cell
1.0
2.5268502235 0.0000000000 0.0000000000
1.2634251118 2.1883164851 0.0000000000
1.2634251118 0.7294388284 2.0631645680
C
2
Direct
0.500000000 0.500000000 0.500000000
0.750000000 0.750000000 0.750000000
- Script for VASP run (name:job.sh):
1
2
3
4
# yhcontrol show job
yhrun -N 1 -n 24 -p work vasp_std > vasp.log 2>&1
>> Submit the task:
1 | yhbatch -N 1 -p work ./job.sh |
- When the calculation is complete, check the log file to make sure the wave function has been written:
>> Create a new folder for calculating band structure of diamond:
mkdir band&cd band&cp ../* ./, then change the parameters of the INCAR file, e.g.:1
2
3
4
5
6
7
8
9
10
11
12
13System = Band
PREC = N
ISTART = 1 ##
ICHARG = 11 ##
ENCUT = 520
EDIFF = 1e-5
EDIFFG = -0.001
IBRION = -1
ISMEAR = 0
SIGMA = 0.05
LCHARG = F ##
LWAVE = F ##
NPAR = 4Create a KPOINGTS (brillouin zone path) file for calculating band structure of diamond. Here, I strongly recommend using the Materials Cloud platform to complete the k point path. Or you can manually set up the k-point file based on this Paper.

In diamond case, we chose the k point path according to the materials cloud. e.g. (file name: KPOINTS):1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24K-Points
20
Line-Mode
Reciprocal
0.00000000 0.00000000 0.00000000 ! G
0.50000000 0.00000000 0.50000000 ! X
0.50000000 0.00000000 0.50000000 ! X
0.62500000 0.25000000 0.62500000 ! U
0.62500000 0.25000000 0.62500000 ! U
0.37500000 0.37500000 0.75000000 ! K
0.37500000 0.37500000 0.75000000 ! K
0.00000000 0.00000000 0.00000000 ! G
0.00000000 0.00000000 0.00000000 ! G
0.50000000 0.50000000 0.50000000 ! L
0.50000000 0.50000000 0.50000000 ! L
0.50000000 0.25000000 0.75000000 ! W
0.50000000 0.25000000 0.75000000 ! W
0.00000000 0.00000000 0.00000000 ! X
>> Submit the task:
1 | yhbatch -N 1 -p work ./job.sh |
- When the calculation is complete, check the log file to make sure the wave function has been written:
>> Plot the band structure using P4VASP:
- Download the
vasprun.xmlfile from the cluster and import this into the P4VASP program: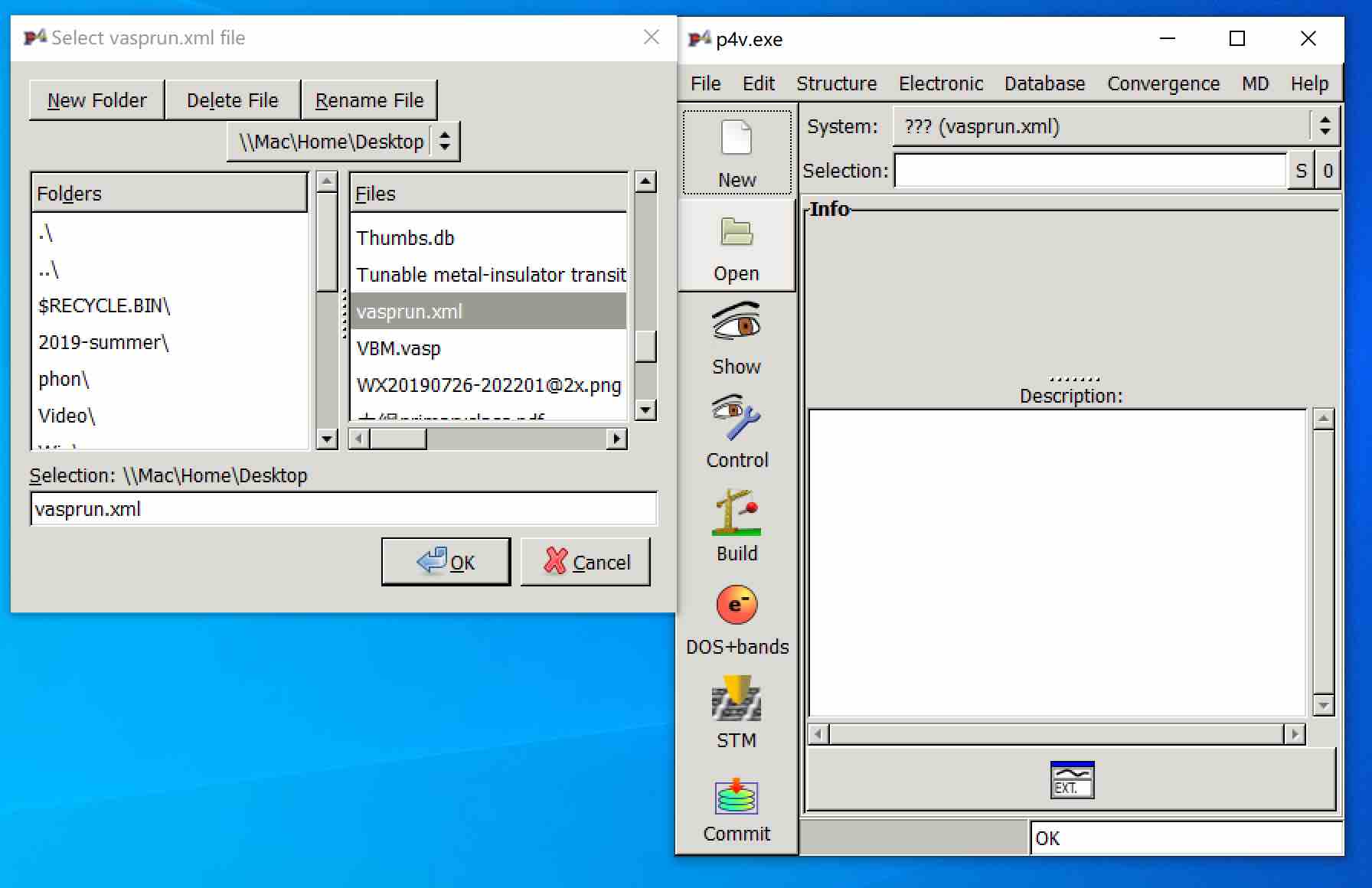
- Click
electronic -> DOS+bands
- Click
Show -> Bands
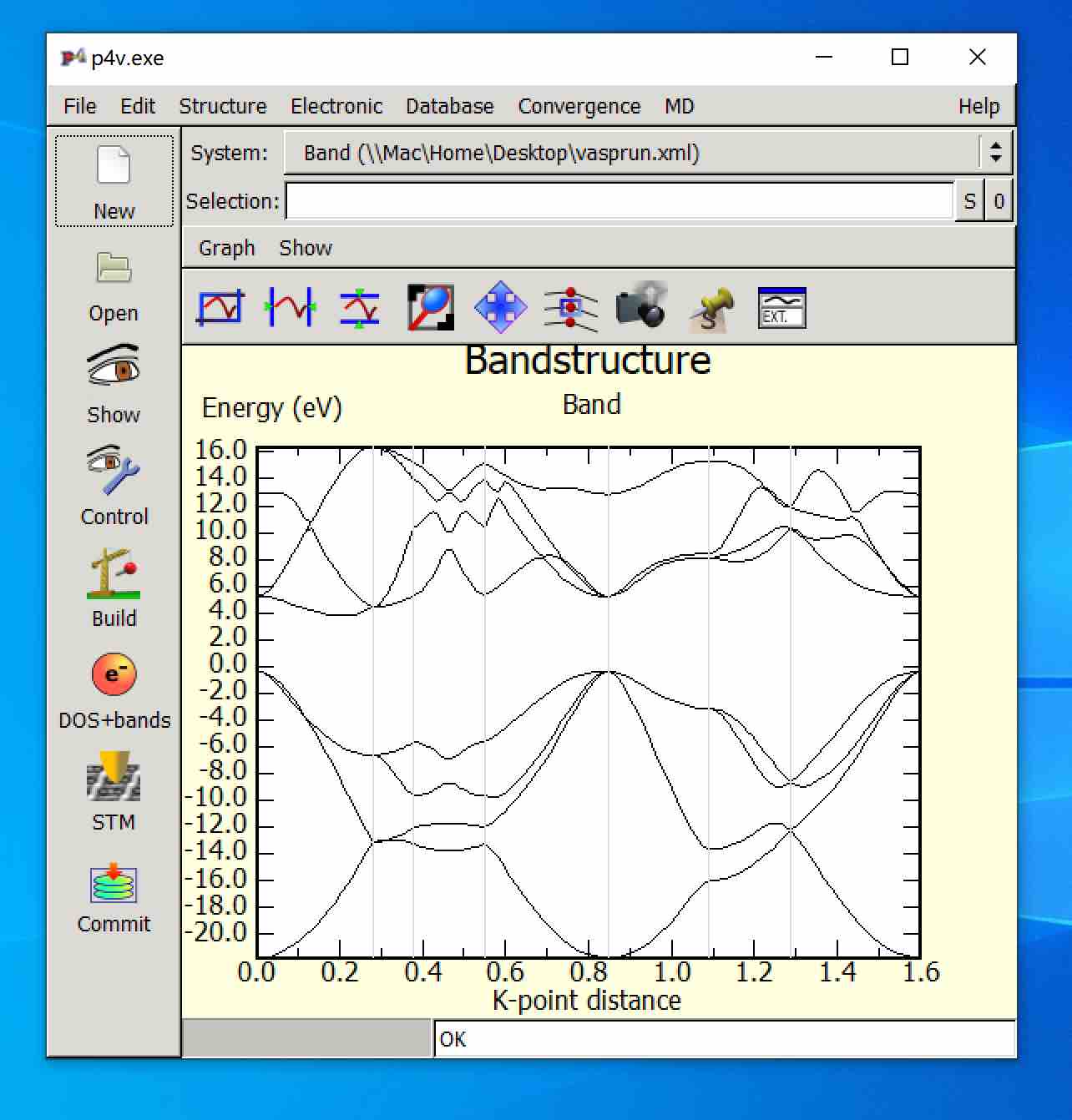
- You will see:
- Export
.datfile: ClickGraph -> Export
- Drag the
.datfile into the Origin: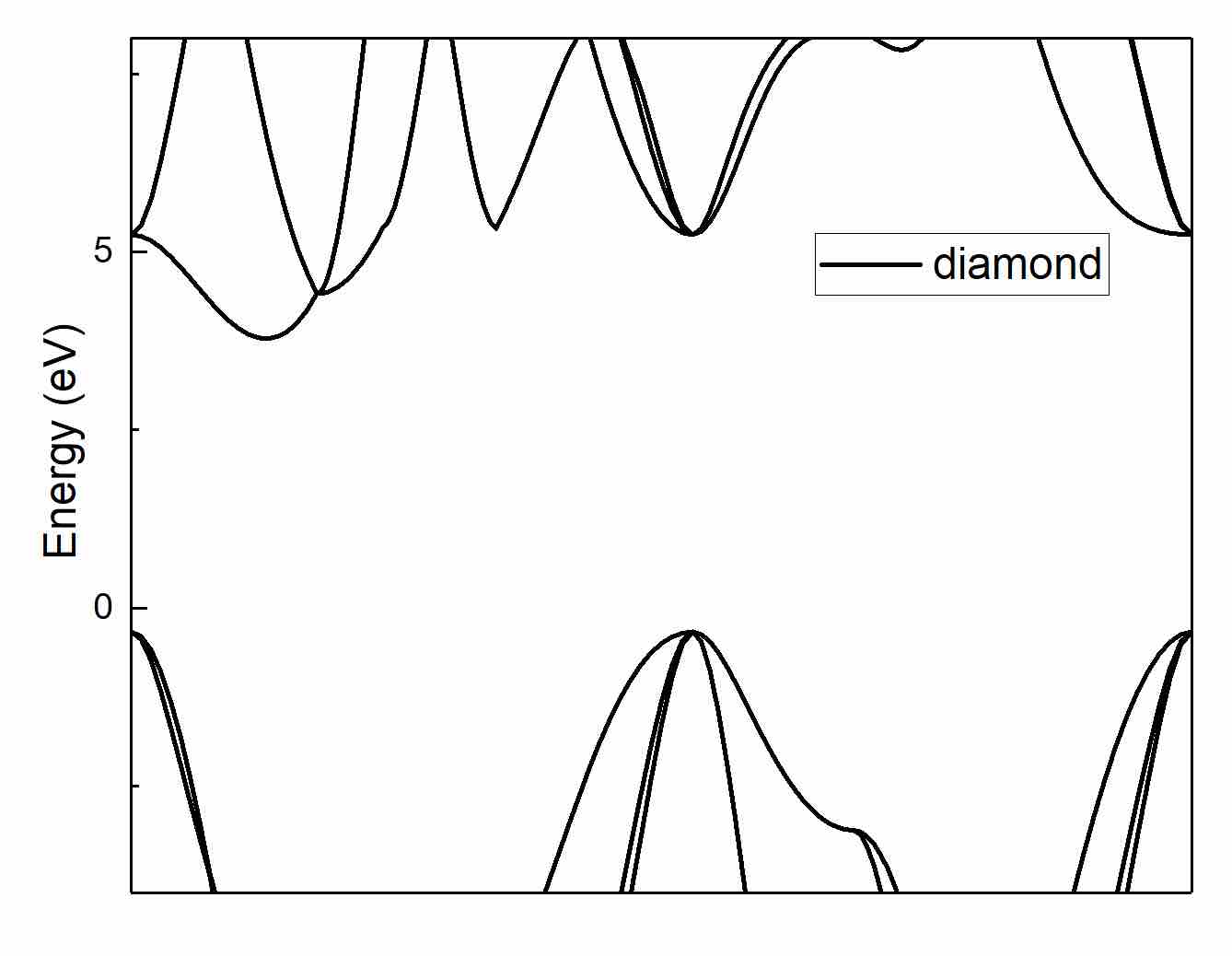
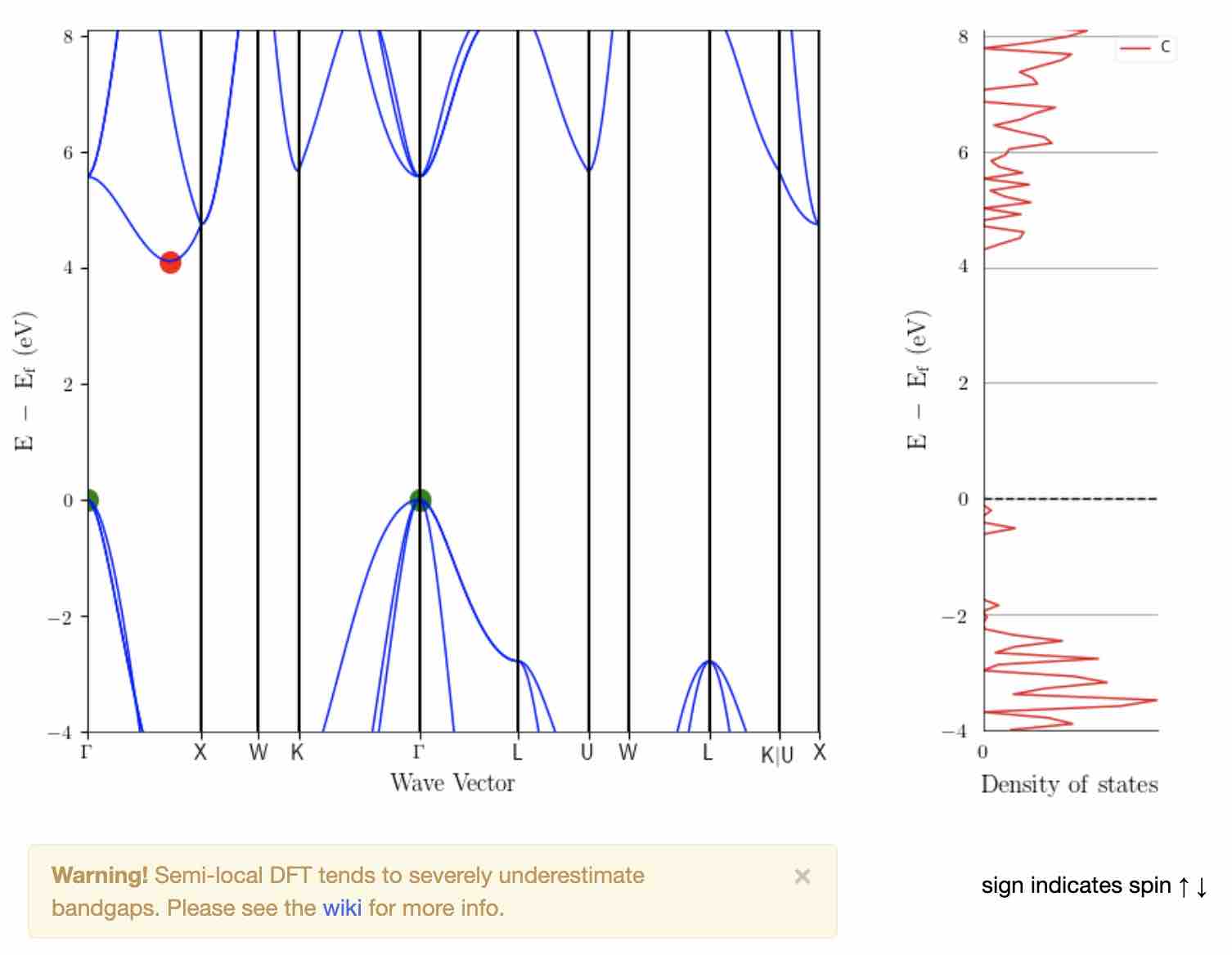
- Comparison with Materials Project database
>> Create a new folder for calculating density of states of diamond:
In
scffolder, creat dos a folder:mkdir dos, and copy all files in scf folder into dos folder:cp ./* ./dos/. Then, go todosfolder:cd ./dos/Change the parameters of the INCAR file, e.g.:
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15System = DOS
PREC = N
ISTART = 1
ICHARG = 11
ENCUT = 520
EDIFF = 1e-5
EDIFFG = -0.001
IBRION = -1
ISMEAR = -5. ##
# SIGMA = 0.05
LCHARG = F
LWAVE = F
NPAR = 4
NEDOS = 2000
LORBIT = 11 # or 10 for per atomChange the
KPOINTSfile, It’s denser than scf calculation, e.g.:1
2
3
4
5K-Mesh
0
Monkhorst-Pack
25 25 25
0.0 0.0 0.0
>> Submit the task:
1 | yhbatch -N 1 -p work ./job.sh |
When the calculation is complete, check the log file:
Plot the pdos using P4VASP:
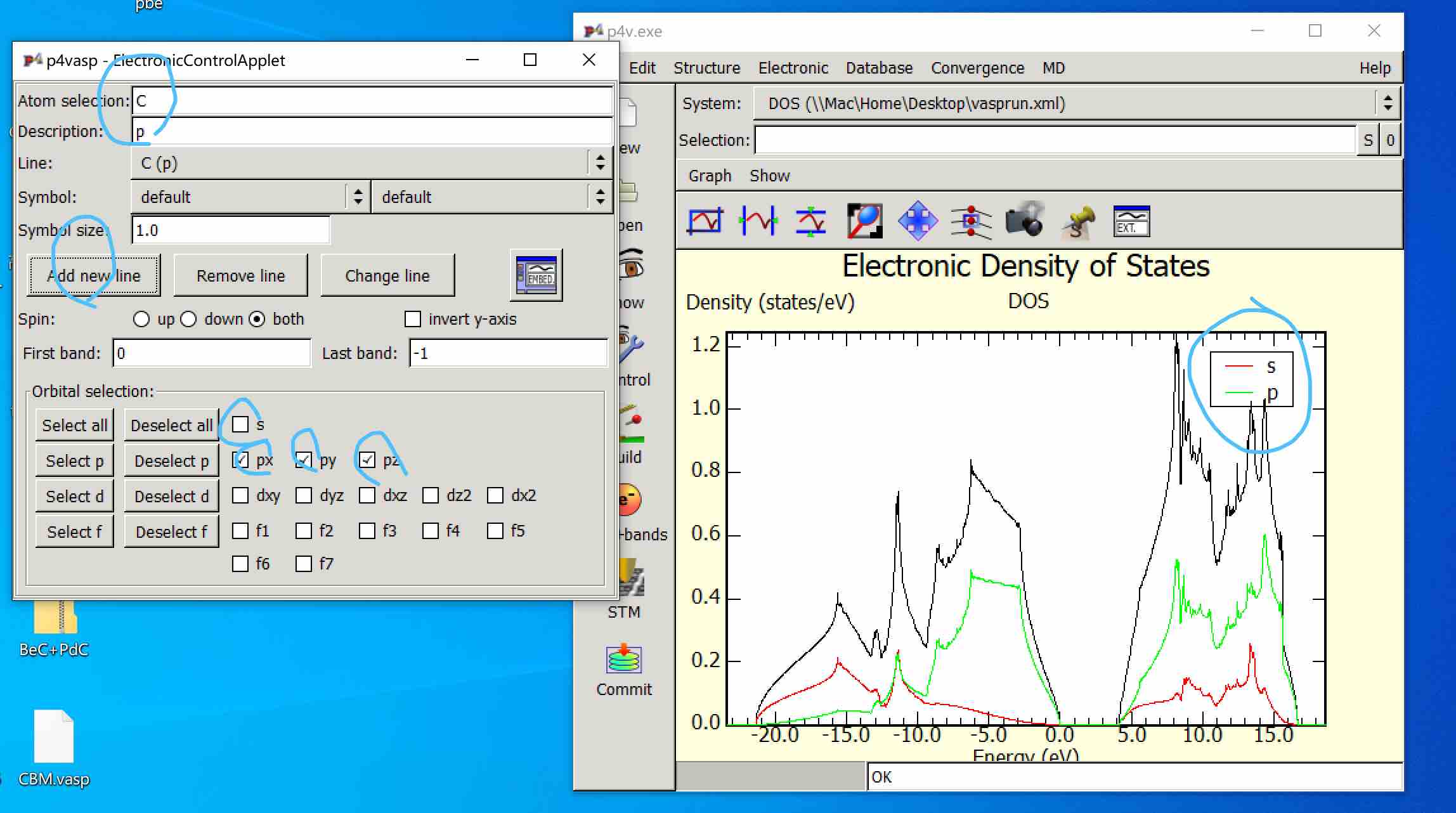
Actually, there are many ways to plot the results, and the best way to do that is to use the shell or python script to extract the data, but using the official P4VSP program is a good choice for beginners.
5. Homework
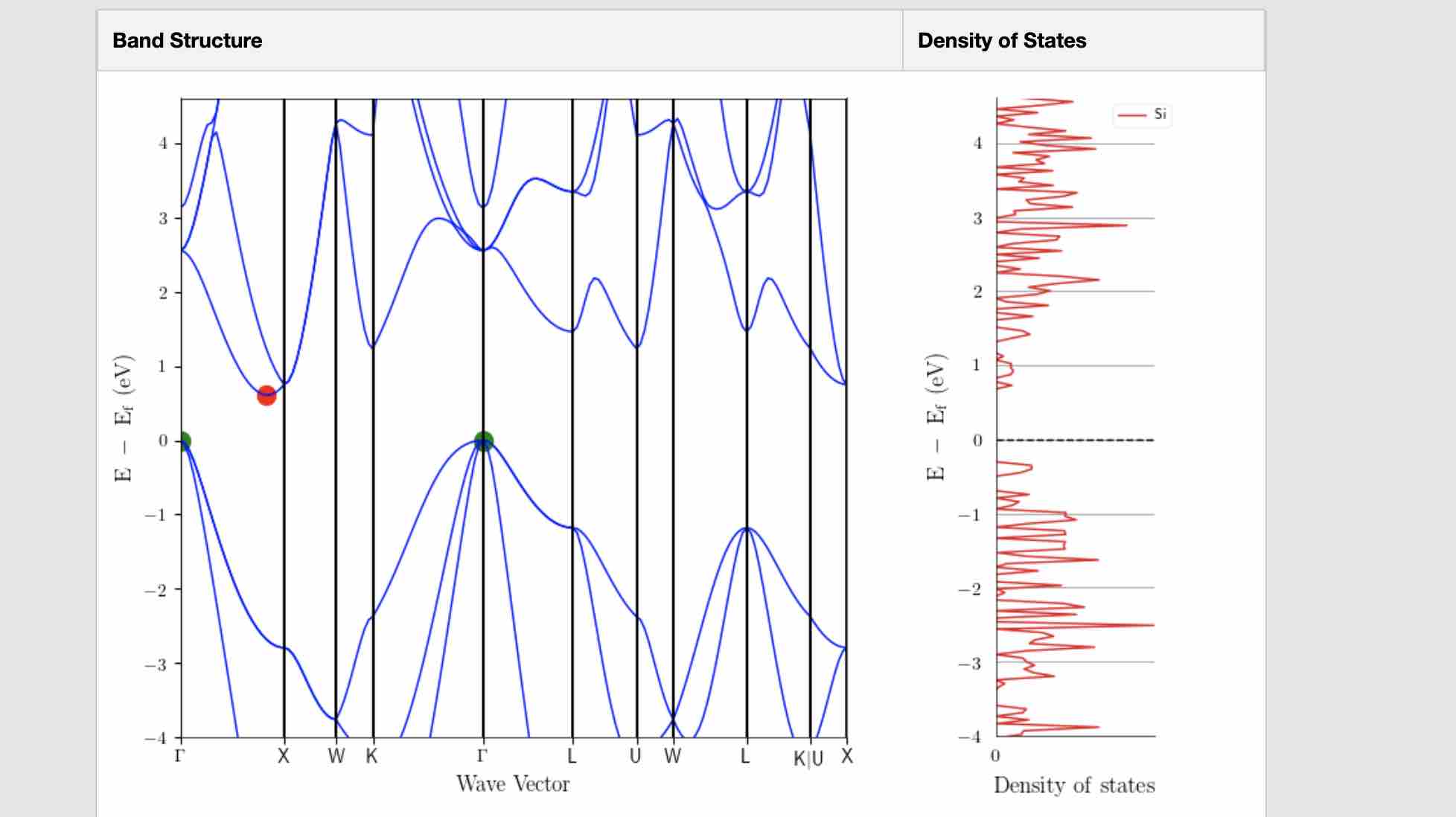
- Calculate band structure and DOS of silicon, Materials Project, MP-149
References
[1] https://www.materialsproject.org
[2] https://www.materialscloud.org
[3] http://www.p4vasp.at
- Blog Link: http://agrh.github.io/2019/07/28/band-diamond/
- Copyright Declaration: The author owns the copyright, please indicate the source reproduced.